
Идентификация той или иной формы синдрома дисплазии соединительной ткани (ДСТ) затруднена в связи с полисистемностью клинических проявлений, наличием стертых и перекрестных форм, а также отсутствием критериев диагностики ввиду гетерогенности (точковая мутация de novo, гетерозиготность, пенетранность, мультифакториальность и т.д.).
Одним из критериев постановки синдрома Марфана является подвывих хрусталика (Ectopia lentis). Вместе с тем Ectopia lentis может быть самостоятельным заболеванием при мутации гена ADAMTSL4 [1].
В настоящий момент ни в зарубежной, ни в отечественной литературе указаний на мутации гена ADAMTSL4 при марфаноподобном фенотипе (марфаноидной внешности) мы не нашли, что подтверждает сложность постановки диагноза. Поэтому поиск новых методов диагностики для подтверждения формы ДСТ крайне актуален.
Целью нашего клинического наблюдения стал поиск генетических мутаций и полиморфизмов, которые могли бы лежать в основе формирования марфаноподобного фенотипа (марфаноидной внешности) у пациентки с несостоятельности рубца на матке после кесарева сечения при ДСТ.
Нами была обследована пациентка 28 лет с марфаноподобным фенотипом (марфаноидной внешностью). Для диагностики синдрома ДСТ использовалась разработанная нами панель для выявления генетических мутаций при ДСТ на основе метода высокопроизводительного секвенирования (NGS). Благодаря ей возможен анализ по точкам: B4GALT7, BMP1, C1R, COL1A1, COL1A2, COL11A1, COL11A2, COL3A1, COL5A1, COL5A2, ELN, FBLN4, FBLN5,FBN1, FBN2, FGFR3, FLNA, MYH11, PLOD1, TGFB1, TGFBR1, TGFBR2, TNXB+ ACTA2, ADAMTS10, ADAMTS2, ADAMTSL4, COL2A1, DCHS1, LOX, MMVP1, MMVP3, MYLK, SMAD3, COL1A1 rs1800012, TGFBrs1800471, COL3A1 rs1800255, FBLN5 rs2018736, FBLN5 rs12589592, COL2A1 rs2276455, COL2A1 rs63118460 (rs7963636), MMP10 rs17435959, MMP10 rs17293607, ESR1 rs2228480, PGRrs484389, MMP13 rs2252070, GDF5 rs143383, MMP3 rs35068180, MMP3 rs3025058, VEGFArs699947, VEGFA rs2010963, VEGFA rs3025039, MMP9 rs17576, MMP9 rs3918242, LAMC1 rs10911193.
ОПИСАНИЕ КЛИНИЧЕСКОГО СЛУЧАЯ
Пациентка, 34 лет, поступила в клинику в мае 2019 г. с диагнозом «несостоятельность рубца на матке после двух кесаревых сечений в анамнезе» (2014 и 2017). Состояние после герниопластики с установкой сетчатого импланта по поводу пупочной грыжи и диастаза мышц переднебрюшной стенки в 2019 г. Спаечный процесс в малом тазу.
В анамнезе: росла и развивалась нормально. Менструации с 12 лет, установились сразу по 5 дней, через 28, умеренные, безболезненные. В анамнезе 2 беременности, 2 родов.
Первые (2014) своевременные оперативные роды – в головном предлежании в сроке 41 нед. Экстренное чревосечение по Пфанненштилю, кесарево сечение в нижнем маточном сегменте было выполнено в связи с развитием первичной слабости родовой деятельности, неэффективностью родоактивации окситоцином, амниотомии. Пациентка разрешилась крупным плодом (4000 г/53 см). Ранний послеродовой период осложнился гипотоническим кровотечением, проводилась консервативная терапия. Выписана на 7-е сутки с уровнем гемоглобина 60 г/л.
Вторые (2017) своевременные оперативные роды – в головном предлежании путем кесарева сечения в плановом порядке в связи с рубцом на матке после кесарева сечения в анамнезе. Вес плода – 4050 г, рост – 55 см.
Анамнез заболевания: через полгода после вторых родов с помощью УЗИ была выявлена несостоятельность рубца на матке. На УЗИ в области рубца на матке после кесарева сечения определялось анэхогенное включение 0,9×0,7 см («ниша»). МРТ подтвердило диагноз: рубец истончен до 0,2 см с формированием на протяжении 1,7 см «ниши». В «нише» – скопление неоднородной жидкости геморрагического характера.
Наследственные заболевания: у отца пациентки язвенная болезнь желудка и двенадцатиперстной кишки.
Жалобы при поступлении: периодически кровяные выделения из половых путей.
Сопутствующие заболевания: с пубертатного возраста пациентка страдает артериальной гипотензией (артериальное давление может снижаться до 90/60 мм рт.ст.). В 2018 г. оперирована по поводу пупочной грыжи. При МРТ обнаружены грыжи межпозвоночных дисков L4–5, L5–S1, начальные проявления спондилеза, гемангиома в теле L 5, периневральные кисты в позвоночном канале на уровне S1–2.
В 2019 г. пациентка повторно подверглась операции в связи с диастазом прямых мышц живота (6 см) в объеме: ей была выполнена герниопластика с установкой сетчатого эндопротеза «УЛьтрапро».
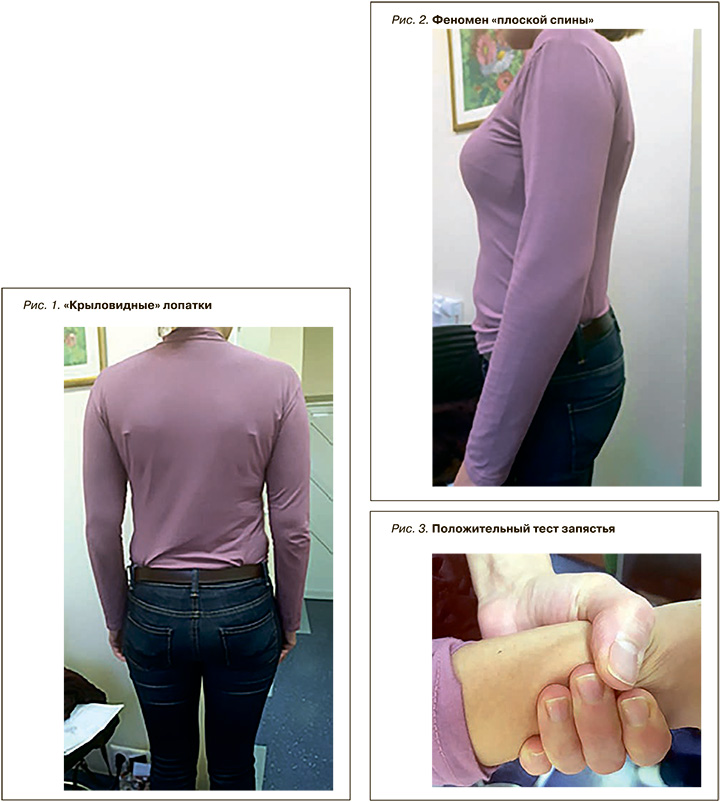
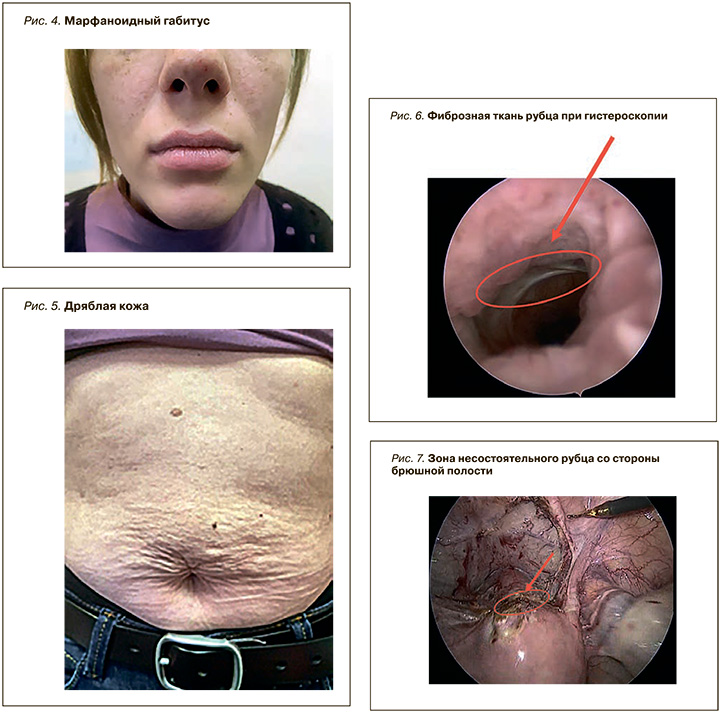
Данные объективного обследования: астеническое телосложение, рост 164, вес 58 кг. Осанка ослаблена, лопатки «крыловидные» (рис. 1), феномен «плоской спины» (рис. 2), размах рук/рост 171/164 см, расстояние от акромиона до кончика третьего пальца руки – 71 см, расстояние в положении стоя от верхнего края лобкового симфиза до пола – 84 см, длина кисти – 19 см, тест запястья положительный (рис. 3), сколиоз 1–2 степени. Наблюдается специфичный Gabitus (рис. 4).
Отмечается умеренное повышение эластичности кожи. Кожа ранимая, на переднебрюшной стенке дряблая, морщинистая (рис. 5).
Выявлено снижение индекса мышечной массы, гипермобильность суставов (только локтевых) – 2 балла.
Результаты общеклинического обследования:
- эхокардиография (ЭхоКГ): пролапс митрального клапана (ПМК) 1 степени 0,3 см, конечно-диастолический объем (КДО) 93 мл, фракция выброса (ФВ) 79%, фракция укорочения (ФУ) 48%, толщина межжелудочковой перегородки (МЖП) 8 мм, толщина задней стенки левого желудочка (ЗСЛЖ) 9 мм, незначительная дилятация левого предсердия в длину (33×52), пролапс трикуспидального клапана (ПТК), митральная регургитация (МР) 1 степени, пролапс аортального клапана (ПАК) 1 степени, аортальная регургитация (АР) 1 степени;
- электрокардиография (ЭКГ): синусовый ритм, 70 уд./мин, нарушение внутрижелудочковой проводимости, неполная блокада правой ножки пучка Гиса;
- допплерометрия вен нижних конечностей: ретикулярный варикоз притоков обеих больших подкожных вен и заднемедиальных коммуникативных анастомозов обеих голеней без патологического рефлюкса.
Интраоперационно подтвержден диагноз несостоятельности рубца на матке (рис. 6 и 7). Выполнена операция: лапароскопия, гистероскопия, рассечение спаек в малом тазу. Произведено иссечение дефекта послеоперационного рубца на матке, метропластика.
При гистологическом исследовании зоны несостоятельности рубца обнаружена фиброзная ткань.
При выполнении анализа на генетические мутации при ДСТ с использованием разработанной нами панели на основе метода высокопроизводительного секвенирования (NGS) выявлен редкий вариант мутации в гене ADAMTSL4.
ОБСУЖДЕНИЕ
В пользу марфаноподобного фенотипа марфаноподобной внешности в рассмотренном клиническом случае говорят клинико-метрические признаки поражения соединительной ткани. Среди костных критериев следует выделить:
- долихостеномелию – отношение длины кисти к росту 11,6%, соотношение длины стопы к росту 15,6%, разность между величинами размаха рук и ростом 7 см;
- арахнодактилию – положительный тест запястья и скрининг большого пальца кисти, «плоская спина» и «крыловидные лопатки», марфаноподобный габитус;
- грыжи межпозвоночных дисков L4–5, L5–S1, начальные проявления спондилеза по данным МРТ.
Среди других критериев на синдром ДСТ указывали дряблая (вялая) кожа, пупочная грыжа в анамнезе, кариес, мышечная гипотрофия, слабость дистальных мышц, ослабленная осанка, диастаз мышц переднебрюшной стенки, приведший к необходимости хирургической коррекции с установкой сетчатого протеза, первичная слабость родовой деятельности, кровотечение в ранний послеродовый период, обусловленный гипотонией матки, ретикулярный варикоз нижних конечностей.
Пациентка с помощью указанной панели для выявления генетических мутаций при ДСТ была обследована на синдром Марфана, а также другие формы ДСТ (синдром Элерса–Данлоса, недифференцированные формы ДСТ). Дефектный ген фибриллина, а также мутации в генах, отвечающих за другие моногенные формы ДСТ, идентифицированы не были.
Тем не менее, несмотря на то что у больной отсутствовал синдром Марфана, а также несиндромная форма подвывиха хрусталика (Ectopia lentis), мутация гена ADAMTSL4 была идентифицирована.
Белки ADAMTSL4 в большом количестве присутствуют в фибриллярных структурах экстрацеллюлярного матрикса других частей глаза, а также, помимо специализированных глазных структур, в стенке артерий (здесь фибриллин 1 типа был представлен в изобилии) [2].
Согласно результатам проведенного нами анализа и по данным литературы, ген ADAMTSL4, а также семейство белков ADAMTS опосредованно ответственны за развитие симптомов и синдромов, которые имеют крайне высокий коэффициент диагностики и информативности в постановке диагноза ДСТ. Согласно клиническим рекомендациям Российского научного медицинского общества терапевтов (РНМОТ) к таким симптомам относятся вентральные грыжи 9,55 и 0,424 соответственно, миопия 6,48 и 1,120, отслойка сетчатки 9,86 и 0,952, трахеобронхиальная дискинезия 6,76 и 1,093 и т.д. [3]. В связи с этим становится понятным появление подобных клинических маркеров ДСТ в нашем клиническом случае при мутации в гене ADAMTSL4.
ЗАКЛЮЧЕНИЕ
Изменение экспрессии белка ADAMTSL4 может быть еще одним из механизмов нарушения микрофибриллогенеза. В эксперименте было показано, что среда, содержащая человеческий ADAMTSL4, ускоряла отложение FBN1 (фибриллина) в микрофибриллах фибробластами связки [2]. Соответственно снижение экспрессии ADAMTSL4 могло бы привеcти к нарушению отложения Fbn1 (фибриллина) в микрофибриллах.
Отсюда можно сделать вывод: как синтез мутантного FBN1 (синдром Марфана), так и нарушения отложения FBN1 (при мутации в гене ADAMTSL4) тормозят образование нормальных микрофибрилл нормальным фибриллином 1 или стимулируют протеолиз несоответствующих внеклеточных микрофибрилл. Указанный механизм вполне может быть причиной формирования марфаноподобного фенотипа (марфаноподобной внешности).


