
Стероидорезистентный нефротический синдром (СРНС) – гетерогенная группа гломерулопатий, характеризующихся резистентностью к терапии высокими дозами глюкокортикоидов (ГК) в течение 8 нед у детей [1]. По данным Союза педиатров России, ежегодная частота возникновения нефротического синдрома составляет 2–7 первичных случаев на 100 000 детской популяции, распространенность 12–16 случаев на 100 000 [1]. В современной классификации выделяют первичный (идиопатический) и вторичный нефротический синдром (генетически обусловленный, ассоциированный с вирусами и другими инфекционными агентами, в составе системных заболеваний и др.) [1].
Генетически обусловленные причины СРНС многообразны: мутации ACTN, NPHS1, NPHS2, CD2AP и др. Мутации гена подоцина NPHS1 и NPHS2 приводят к развитию подоцитопатии, в результате которой нарушается структура и функция «щелевой» диафрагмы, что приводит к повышению ее проницаемости для крупных белковых молекул [2].
Мутация гена NPHS1 сопровождается развитием врожденного нефротического синдрома по финскому типу, когда наблюдается внутриутробная протеинурия и нефроз после рождения с возможным последующим развитием врожденной почечной недостаточности. Продолжительность жизни при этой патологии составляет около года [2–5].
Мутация гена NPHS2 клинически проявляется стероидорезистентной формой нефротического синдрома у детей, имеющей более благоприятный прогноз. Дебют заболевания наблюдается в возрасте от 7 до 12 лет. Мутация гена подоцина NPHS2 имеет аутосомно-рециссивный тип наследования, при этом СРНС возникает не менее чем у 2 членов одной семьи, чаще у сибсов. Морфологические изменения в почках представлены преимущественно фокальным сегментарным гломерулосклерозом (ФСГС) и болезнью минимальных изменений. Отмечена низкая частота возврата ФСГС после трансплантации почки (3–8%) [2–5].
КЛИНИЧЕСКОЕ НАБЛЮДЕНИЕ № 1
Пациент А., 20 лет. Впервые в марте 2006 г. (7 лет) выявлен развернутый нефротический синдром (суточная протеинурия 15 г, гипопротеинемия 49 г/л, гипоальбуминемия 24 г/л, гиперхолестеринемия 12,2 ммоль/л, отеки). Повышение креатинина, анемия и артериальная гипертензия не регистрировались. В ходе обследования был исключен вторичный генез нефротического синдрома (вирусные гепатиты, опухоли, системные заболевания). Выставлен диагноз: «хронический гломерулонефрит, нефротическая форма, обострение».
Пациенту проводилась программная активная иммуносупрессивная терапия: пульс-терапия преднизолоном по 500 мг трижды, преднизолон из расчета 1 мг/кг/сут внутрь в течение 8 нед с последующим переводом на альтернирующую схему приема этого ГК – 1,5 мг/кг/48 ч в течение 4 нед. Также больной получал неспецифическую нефропротективную терапию (ингибиторы АПФ, дезагреганты, статины), мочегонные препараты (фуросемид). Эффекта от проводимого лечения не было, в связи с чем нефротический синдром был расценен как стероидорезистентный.
В августе 2006 г. в «Национальном медицинском исследовательском Центре здоровья детей» (Москва) пациенту была выполнена нефробиопсия. Заключение по результатам исследования: ФСГС.
К лечению был добавлен циклоспорин по 3 мг/кг массы тела, терапия проводилась в течение полугода, доза препарата титровалась в зависимости от концентрации в сыворотке крови. Эффекта от проводимого лечения также не было.
В марте 2007 г. было проведено генетическое исследование, по результатам которого у пациента выявили гомозиготную мутацию гена подоцина NPHS2. С учетом полученных данных был выставлен диагноз: «СРНС, обусловленный мутацией гена подоцина NPHS2, морфологически вторичный ФСГС». Осложнения: медикаментозный синдром Иценко–Кушинга.
Иммуносупрессивная терапия была постепенно отменена, продолжены неспецифическая нефропротекция (ингибиторы АПФ, статины) и симптоматическое лечение. С течением времени отмечалось закономерное развитие и прогрессирование симптомов хронической почечной недостаточности (появление артериальной гипертензии, анемии), выполнялась их коррекция.
Впервые в отделение нефрологии Областной клинической больницы Саратова пациент был госпитализирован в 2016 г. Обращала внимание задержка у него физического развития, что трактовалось как осложнение длительной терапии ГК в раннем школьном возрасте. В ходе обследования была констатирована 5 стадия хронической болезни почек, начата заместительная почечная терапия перитонеальным диализом. В 2017 г. (19 лет) выполнена трансплантация почки от матери, функция трансплантата срочная, назначена комбинированная иммуносупрессивная терапия (такролимус, препараты мофетилмикофенолатов, глюкокортикоиды). В сентябре 2018 г. нарушения функции трансплантата не выявлено.
КЛИНИЧЕСКОЕ НАБЛЮДЕНИЕ № 2
Пациент С., 18 лет, родной брат пациента А. Впервые изолированная протеинурия (2 г в сутки) была выявлена в марте 2007 г. (7 лет). С учетом отягощенной наследственности было выполнено генетическое исследование, по итогам которого у пациента выявлена гомозиготная мутация гена подоцина NPHS2, у родителей – гетерозиготные мутации гена подоцина NPHS2. Основываясь на полученных данных и отсутствии эффекта от иммуносупрессивной терапии у родного брата, пациенту была назначена неспецифическая нефропротективная терапия – ингибиторы АПФ, дезагреганты.
В декабре 2007 г. в «Национальном медицинском исследовательском Центре здоровья детей» (Москва) пациенту была выполнена нефробиопсия: заключение «мезангиопролиферативный гломерулонефрит».
С 2013 г. (13 лет) отмечалось появление артериальной гипертензии (максимальное повышение артериального давления (АД) до 150 и 100 мм рт.ст.), в связи с чем пациенту была увеличена доза ингибитора АПФ.
В 2014 г. (14 лет) у пациента впервые был зарегистрирован развернутый нефротический синдром (общий белок 36 г/л, альбумины 13 г/л, холестерин 11 ммоль/л, протеинурия 5 г/л, отеки), креатинин 35 мкмоль/л, гемоглобин 135 г/л. В 2015 г. больной находился на обследовании в «Национальном медицинском исследовательском Центре здоровья детей» (Москвы), ему было рекомендовано продолжить неспецифическую нефропротективную терапию.
Впервые в отделение нефрологии Областной клинической больницы Саратова пациент госпитализирован в сентябре 2018 г. При поступлении предъявлял жалобы на отеки ног до верхней трети голеней, головные боли в затылочной области и носовые кровотечения, возникающие при повышении АД до 150/100 мм рт.ст. Данные объективного осмотра: физическое развитие соответствует возрастной норме, отеки нижних конечностей до средней трети бедер, АД 150/90 мм рт.ст., диурез 1,5 л в сутки. По другим органам без особенностей.
Общий анализ крови: гемоглобин 115 г/л, эритроциты 3,5×1012/л, лейкоциты 7,8×109/л, тромбоциты 320×109/л, СОЭ 53 мм/ч. Биохимический анализ крови: общий белок 46 г/л, альбумин 16 г/л, холестерин 8,2 ммоль/л, мочевина 8 ммоль/л, креатинин 102 мкмоль/л.
Общий анализ мочи: удельный вес 1010, белок 3 г/л, мочевого осадка нет. Суточная протеинурия 3,3 г/л. В динамике отмечалось появление анемии и нарастание уровня креатинина.
Ультразвуковое исследование почек: размеры почек в норме, толщина паренхимы 15 мм с обеих сторон, умеренно повышена эхогенность паренхимы.
Дуплексное исследование почечных артерий и вен: патологии не выявлено.
Эхокардиография: изменений структуры и функции миокарда, клапанного аппарата сердца не выявлено. Размеры сердца: индекс массы миокарда 72 г/м, толщина задней стенки левого желудочка 0,85 см, фракция изгнания левого желудочка 73%.
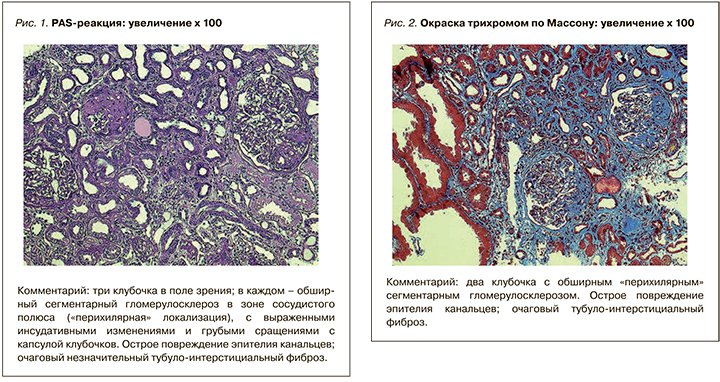
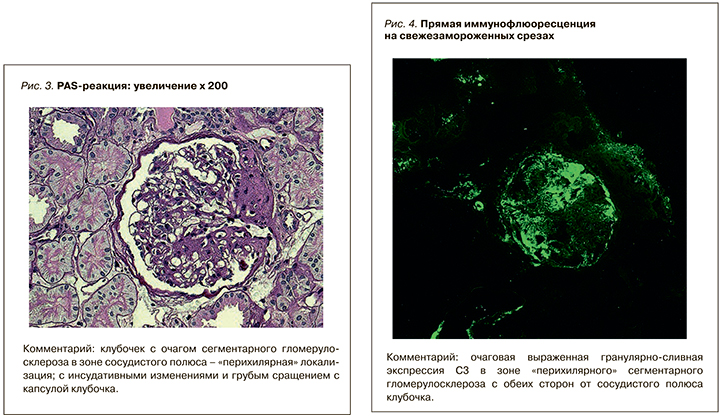
В сентябре 2018 г. пациенту выполнена повторная нефробиопсия для оценки степени выраженности склероза. Материал биопсии исследован в «Национальном центре клинической морфологической диагностики» (Санкт-Петербург), по результатам которого вынесено следующее заключение: «гистологическая картина вторичного обширного „перихилярного” сегментарного гломерулосклероза (60%), ассоциированного с верифицированной мутацией гена NPHS2 и дефицитом подоцина; полный гломерулосклероз (17%); незначительный тубулоинтерстициальный фиброз (20%); умеренный артериолосклероз» (рис. 1–4).
Учитывая отягощенный семейный анамнез и данные дополнительных методов обследования, пациенту был выставлен диагноз: «семейный СРНГ, обусловленный мутацией гена подоцина NPHS2, морфологически вторичный ФСГС. Вторичная артериальная гипертензия». Осложнения: нефрогенная анемия легкой степени.
В связи с генетическим характером заболевания, отсутствием эффекта на фоне комбинированной иммуносупрессивной терапии у родного брата основными целями лечения этого пациента было отдаление сроков наступления терминальной стадии заболевания почек (максимальная нефропротективная терапия, симптоматическое лечение нефротического синдрома) и профилактика инфекционных заболеваний. Ему было рекомендовано продолжить прием фозиноприла (20 мг/сут), аторвастатина (40 мг/сут), ацетилсалициловой кислоты (100 мг/сут), торасемида (10 мг/сут), железа сульфата + аскорбиновой кислоты (1 табл. в день). На фоне постоянного приема препаратов отмечалась положительная динамика – уменьшение отечного синдрома, стабилизации цифр АД.
ЗАКЛЮЧЕНИЕ
Представленные клинические наблюдения демонстрируют семейный вариант СРНС, обусловленного мутацией гена подоцина NPHS2. Характерные черты СРНС в представленных наблюдениях включают дебют заболевания в возрасте 7 лет у обоих сибсов, безуспешность иммуносупрессивной терапии с развитием осложнений (задержка физического развития), констатация терминальной стадии заболевания почек спустя 11 лет от начала заболевания, успешная родственная нефротрансплантация у старшего брата. Особенности СРНС у младшего брата – сохранная функция почек в течение 12 лет от начала заболевания на фоне неспецифической нефропротективной терапии.
Согласно данным литературы, иммунносупрессивная терапия при СРНС, обусловленном мутацией гена подоцина NPHS2, не показана. Наше клиническое наблюдение демонстрирует безуспешность такого лечения, развитие осложнений на фоне иммуносупрессивной терапии и более раннее наступление терминальной стадии заболевания почек. Основными направлениями терапии у данной группы пациентов следует признать максимальную нефропротекцию (назначение ингибиторов АПФ или сартанов, статинов, дезагрегантов), при необходимости симптоматическое лечение (мочегонные препараты, трансфузия альбумина, антикоагулянты). Следует подчеркнуть важность динамического наблюдения за пациентами: контроль АД, биохимических показателей крови (общего белка, альбумина, креатинина, суточной протеинурии, расчет СКФ 1 раз в 2 мес), общего анализа крови, суточной протеинурии с целью своевременной коррекции проявлений хронической почечной недостаточности и планирования заместительной почечной терапии.
Применение генетического исследования у пациентов с СРНС целесообразно выполнять у детей. Также возможно его проведение у пациентов с впервые выявленным нефротическим синдромом, начальными признаками почечной недостаточности, отягощенным семейным анамнезом в возрасте 16–20 лет, так как наследственный характер заболевания определяет тактику ведения пациента.


