
Согласно результатам эпидемиологических исследований, проведенных в России, распространенность сердечной недостаточности возросла с 1998 г. на 5,3% и достигла 10,2% (14,9 млн пациентов) в 2014 г. [1]. При этом высокий процент больных сердечной недостаточностью (до 40%) имеет нормальную фракцию выброса левого желудочка [1, 2].
Прогноз пациентов, имеющих сердечную недостаточность с сохраненной фракцией выброса (СНсФВ), во многом не отличается от такового у больных со сниженной систолической функцией левого желудочка. По данным американского регистра GWTG-HF, пациенты с сердечной недостаточностью имеют высокий риск смерти в течение года после госпитализации с декомпенсацией заболевания независимо от значения фракции выброса левого желудочка [3]. Долгосрочный прогноз больных СНсФВ также неблагоприятный: в течение 5 лет после госпитализации по причине сердечной недостаточности умирают до 75% пациентов, т.е. такой же процент, как и при сердечной недостаточности с низкой фракцией выброса [4].
В основе СНсФВ лежит диастолическая дисфункция левого желудочка, которая характеризуется повышением конечно-диастолического давления в левом желудочке, в дальнейшем повышением давления в левом предсердии и сосудах малого круга кровообращения (с развитием легочной гипертензии), а затем и в правом предсердии (повышение центрального венозного давления). Такие изменения внутрисердечной гемодинамики определяют появление застоя в почках и печени с нарушением их функции в виде снижения скорости клубочковой фильтрации (СКФ) и гипоальбуминемии, которые встречаются у 50 и 30% пациентов с СНсФВ соответственно [5, 6]. Гемодинамические изменения также могут объяснять появление наиболее частого симптома СНсФВ – снижение толерантности к физическим нагрузкам. Но причиной этого может быть и характерное для данных пациентов снижение выносливости скелетных мышц, в том числе с развитием саркопении в 20% случаев [7]. В настоящем обзоре освещены основные механизмы поражения почек, печени и скелетной мускулатуры при СНсФВ и их связь с развитием этой формы заболевания.
ОСНОВНАЯ ПАРАДИГМА ПАТОГЕНЕЗА СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ С СОХРАНЕННОЙ ФРАКЦИЕЙ ВЫБРОСА
По современным представлениям, в патогенезе СНсФВ основная роль отводится хроническому системному воспалению [8]. У пациентов с СНсФВ, по сравнению с пациентами со сниженной фракцией выброса, наблюдается повышение концентрации таких маркеров системного воспаления, как фактор некроза опухоли-альфа (ФНО- α), С-реактивный белок (СРБ), интерлейкин-6 (ИЛ-6) [9–11]. По данным клинических исследований, увеличение уровня этих провоспалительных цитокинов и СРБ коррелирует с параметрами диастолической дисфункции левого желудочка [12]. К состояниям, которые могут запускать и поддерживать хронический системный воспалительный процесс, относят ожирение, сахарный диабет (СД) 2 типа, артериальную гипертензию (АГ) – заболевания, часто встречающиеся у больных СНсФВ [13]. Хроническое системное воспаление приводит к окислительному стрессу эндотелиальных клеток и дисфункции эндотелия, в том числе в коронарном русле. В процессе дальнейшего иммунологического каскада клеточной трансформации (экспрессия на поверхности эндотелиальных клеток молекул клеточной адгезии (VCAM, E-селектин), миграция моноцитов в субэндотелиальное пространство, трансформация моноцитов в макрофаги и выработка трансформирующего фактора роста β, активация фибробластов) увеличивается синтез и накопление коллагена в миокарде. Это в сочетании со снижением биодоступности оксида азота и активности протеинкиназы G может приводить к гипертрофии, фиброзу, повышению ригидности сердечной мышцы и в итоге к нарушению диастолической функции сердца. Указанные звенья патогенеза являются компонентами основной парадигмы СНсФВ [8].
Хроническое системное воспаление, а также гемодинамические изменения на фоне ремоделирования сердца могут выступать причиной структурных и функциональных изменений других органов – почек, печени, скелетной мускулатуры.
МЕХАНИЗМЫ НАРУШЕНИЯ ФУНКЦИИ ПОЧЕК ПРИ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ С СОХРАНЕННОЙ ФРАКЦИЕЙ ВЫБРОСА
Нарушение функции почек достаточно распространено среди пациентов с СНсФВ. По данным наблюдательного исследования, у 49% больных этой формой сердечной недостаточности наблюдалось снижение СКФ менее 60 мл/мин/1,73м2 [5]. Согласно результатам анализа функции почек, проведенного в исследовании PARAMOUNT, у 62% пациентов имелся как минимум один показатель почечной дисфункции – снижение СКФ или альбуминурия): в 23% случаев наблюдалось только снижение СКФ (менее 60 мл/мин/1,73м2), в 16% была выявлена изолированная альбуминурия (соотношение «альбумин/креатинин в моче» более 30 мг/г), а в 23% отмечалось отклонение обоих показателей [14]. При этом нарушение функции почек в обоих исследованиях было ассоциировано с изменением структурных параметров левого желудочка [5, 14].
Развитие СНсФВ у пациентов с имеющейся хронической болезнью почек (ХБП) наблюдается не так часто. Так, в китайском исследовании с участием 5808 пациентов с диагностированной ХБП (СКФ менее 60 мл/мин/1,73м2, или соотношение «альбумин/креатинин мочи» более 30 мг/г) распространенность СНсФВ составила лишь 2,5% [15]. Стоит отметить, что частота встречаемости СНсФВ возрастает с прогрессированием ХБП: например, она развивается у 10% пациентов на гемодиализе [16].
Для описания взаимосвязи патологии сердца и почек используется термин «кардиоренальный синдром», который был введен в медицинскую литературу Ledoux P. еще в 1951 г. [17]. Но лишь спустя полвека была разработана и принята концепция кардиоренальных взаимодействий и выделены 5 типов кардиоренальных синдромов [18].
Развитие почечной дисфункции при СНсФВ можно отнести к проявлению кардиоренального синдрома типа 2, когда прогрессирующее повреждение почек происходит в результате хронической патологии сердца (в том числе структурного ремоделирования миокарда и нарушения диастолической функции). СНсФВ может возникнуть на фоне имеющейся у пациента ХБП, и в данном случае можно говорить о развитии кардиоренального синдрома типа 4. Наибольший интерес представляет кардиренальный синдром типа 5, при котором, согласно определению, оба органа поражаются одновременно на фоне острого или хронического системного заболевания (СД, АГ, ожирение и др.). Некоторые авторы высказывают мнение, что именно такая концепция наиболее подходит для описания кардиоренальных взаимоотношений у пациентов с СНсФВ и нарушением функции почек. При этом могут различаться степень поражения органов и последовательность манифестации заболеваний, но в основе изменений лежат единые патофизиологические механизмы: хроническое системное воспаление, эндотелиальная дисфункция и фиброз [19].
Стоит отметить, что заболевания, которые могут запускать и поддерживать хроническое системное воспаление (АГ, СД 2 типа, ожирение), служат ведущими причинами развития ХБП, к лабораторным проявлениям которой, независимо от причины, относятся снижение СКФ и альбуминурия [20].
Каскад событий на фоне хронического системного воспалительного процесса, а в дальнейшем окислительного стресса эндотелиальных клеток и дисфункции эндотелия, происходящий в почках, подобен изменениям в миокарде, описанным выше в рамках основной парадигмы СНсФВ: под действием факторов роста, вырабатываемых мигрирующими в субэндотелиальное пространство перитубулярных капилляров моноцитами, происходит активация фибробластов и усиленный синтез коллагена в почках, что приводит к тубулоинтерстициальному фиброзу [21]. Прогрессирующий канальцевый стеноз на фоне тубулоинтерстициального фиброза может приводить к повышению внутриканальцевого давления и в итоге к падению СКФ [22].
Изменения в почках не ограничиваются интерстицием. Подавление синтеза оксида азота как проявление эндотелиальной дисфункции может снижать перфузию и фильтрацию в почке, что, вероятно, связано с вазоконстрикцией афферентной артериолы почечного клубочка [23]. Вследствие нарушения процессов ауторегуляции кровотока в почечном клубочке повышается внутриклубочковое давление; это влечет за собой уменьшение числа подоцитов, истончение базальной мембраны, а в дальнейшем протеинурию [24]. В экспериментальных моделях также показано, что воздействие оксидативного стресса на эндотелиальные клетки способно вызывать разрушение сети гликопротеинов, покрывающей просвет капилляров клубочка между ножками подоцитов, что, в свою очередь, может увеличивать проницаемость гломерулярного фильтра и приводить к альбуминурии [25]. Добавим, что в клиническом исследовании пациентов с СД 2 типа была продемонстрирована связь альбуминурии с показателями эндотелиальной дисфункции [26]. Интересен и тот факт, что такая связь отсутствовала у пациентов без СД – это еще раз подчеркивает роль системного заболевания в запуске патологических механизмов, лежащих в основе СНсФВ и почечной дисфункции.
После дебюта заболевания одного органа (сердца или почек) к патологическому каскаду присоединяются новые звенья, которые усугубляют течение заболевания второго органа и ускоряют его прогрессирование.
Одним из основных патофизиологических механизмов нарушения функции почек при СНсФВ выступает повышение центрального венозного давления (ЦВД) [27]. В клинических исследованиях продемонстрировано, что у пациентов с СНсФВ отмечается повышение давления в правом предсердии и ЦВД в покое [28–30]. Возрастание ЦВД приводит к увеличению давления в эфферентной артериоле почечного клубочка, что, по закону Пуазейля, замедляет кровоток в капиллярах клубочка и снижает фильтрацию [27]. На фоне повышения ЦВД может повышаться внутрипочечное интерстициальное давление, что ведет к интерстициальному фиброзу и снижению фильтрационной функции почек [31].
В основе патогенеза нарушения функции сердца при ХБП лежит задержка воды и натрия в организме в результате активации ренин-ангиотензин-альдостероновой системы при снижении фильтрации. Следствием этого становятся увеличение преднагрузки левого желудочка, нарушение процессов расслабления, гипертрофии миокарда, снижение «податливости» и повышение его ригидности и как итог диастолическая дисфункция. У больных с терминальной стадией ХБП, находящихся на гемодиализе, дополнительное повышение жесткости сосудистой стенки приводит также к увеличению постнагрузки левого желудочка, что усугубляет диастолическую дисфункцию [16]. Немаловажную роль играют и негемодинамические факторы, такие как стимуляция системного воспаления и эндотелиальная дисфункция [16]. Так, в исследовании пациентов с ХБП увеличение маркеров системного воспаления и эндотелиальной дисфункции ассоциировалось с увеличением индекса массы миокарда левого желудочка – одного из диагностических критериев СНсФВ [32].
Таким образом, взаимосвязь нарушения функции сердца при сохранной фракции выброса левого желудочка и почек представляет собой сложную цепочку патофизиологических механизмов, в основе которой могут лежать единые патологические процессы.
МЕХАНИЗМЫ НАРУШЕНИЯ ФУНКЦИИ ПЕЧЕНИ У БОЛЬНЫХ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТЬЮ С СОХРАНЕННОЙ ФРАКЦИЕЙ ВЫБРОСА
Как было описано выше, при СНсФВ часто отмечается повышение ЦВД, что может быть причиной застоя не только в почках, но и печени. Повышение давления в нижней полой вене передается на печеночные синусоиды, в результате чего возникает их дилатация, отек перисинусоидального пространства, тромбоз и геморрагии [33]. В вышеописанных условиях на фоне гипоксии происходит некроз гепатоцитов, прилегающих к центральной вене печеночной дольки [34]. Длительные застойные явления в печени могут вызывать фиброз и появление фиброзных перемычек (септ) между центральными венами печеночных долек [33]. Данные гистологические процессы могут протекать субклинически, проявляясь лишь дискомфортом в правом подреберье. К лабораторным изменениям можно отнести повышение активности печеночных трансаминаз, гамма-глутамилтрансферазы (ГГТ), концентрации билирубина, гипоальбуминемию [33, 34].
Интересные данные, касающиеся концентрации сывороточного альбумина, были получены у пациентов с СНсФВ в исследовании TOPCAT. Было установлено, что гипоальбуминемия (концентрация альбумина крови менее 39 г/л), обнаруженная у 30% пациентов, ассоциируется с повышенным уровнем маркеров системного воспаления (ФНО- α), изменением показателей диастолической дисфункции левого желудочка (Е/е’, индекс объема левого предсердия). Также снижение концентрации сывороточного альбумина было связано с увеличением значения индексов фиброза печени (Fib-4, модифицированный NFS) [6]. Учитывая, что у пациентов в данном исследовании среднее значение СКФ составляло примерно 65 мл/мин, а соотношение «альбумин/креатинин мочи» не превышало 41,8 мг/г, можно предположить, что развитие гипоальбуминемии было обусловлено именно снижением синтеза альбумина в печени, а не повышением его выведения почками, что указывает на наличие печеночной дисфункции (на фоне застойных явлений и, возможно, фиброзных изменений) при СНсФВ.
Поражение печени у больных СНсФВ может быть ассоциировано не только с застойными явлениями, но и метаболическими нарушениями на фоне сопутствующих заболеваний (СД 2 типа, ожирение, в особенности висцеральное ожирение). В этом случае можно говорить о нарушении именно липидного обмена в печени, т.е. о развитии неалкогольной жировой болезни печени (НАЖБП), которая часто встречается у пациентов с данными патологиями (до 75% при СД 2 типа и до 76% при ожирении) [35]. По данным небольшого исследования пациентов с СНсФВ, только у 27% из них была выявлена НАЖБП, в том числе по результатам визуализирующих методик. При этом наличие СД 2 типа и ожирения (ИМТ >30 кг/ м2) было связано с увеличением риска развития НАЖБП при СНсФВ [36].
В настоящее время основной гипотезой, описывающей патогенез НАЖБП, является гипотеза «двойного удара». Согласно ей, под «первым ударом» подразумевается накопление триглицеридов в гепатоцитах (стеатоз печени). «Второй удар» – это неконтролируемая выработка цитокинов (ФНО-α, ИЛ-6 и др.) и активных форм кислорода (оксидативный стресс) в печени, которая приводит к повреждению и гибели гепатоцитов, развитию воспаления и фиброза (стеатогепатит) [35].
Как было описано выше, в развитии СНсФВ важное место занимает эндотелиальная дисфункция коронарного русла. Значение эндотелиальной дисфункции активно изучается и в патогенезе НАЖБП. В ряде работ на животных моделях изучалась роль в развитии НАЖБП дисфункции эндотелиальных клеток сосудистой системы печени (в частности, печеночных синусоидов) [37–40]. Результаты исследований свидетельствуют о том, что уже на ранних стадиях НАЖБП, а именно на стадии стеатоза, в печени снижается биодоступность оксида азота, активность NO-синтазы, повышается выработка активных форм кислорода [37, 40]. Результаты экспериментальных работ подтверждаются данными клинических исследований. Так, у пациентов с НАЖБП было продемонстрировано снижение концентрации функционально активной фракции NO-синтазы в печени по данным иммуногистохимического анализа биоптата в сравнении с группой контроля [41].
Кроме того, на животных моделях было показано, что при стеатозе печени (в отсутствие признаков воспаления и фиброза) происходит активация эндотелиальных клеток синусоидов, на их поверхности увеличивается экспрессия молекул адгезии (ICAM-1, E-селектин и др.), что вызывает активацию и миграцию моноцитов. Также в эндотелиальных клетках, клетках Купфера и звездчатых клетках синусоидов повышается экспрессия мРНК провоспалительных цитокинов (ИЛ-6, ФНО-α) и профибротических факторов [38, 39]. Вышеперечисленные данные позволяют предположить, что дисфункция эндотелиальных клеток синусоидов может играть непосредственную роль в запуске воспаления и фиброза в печени и служить важным связующим звеном в патогенезе НАЖБП.
Для оценки фиброза печени в клинической практике часто используются индексы неинвазивной диагностики фиброза, в том числе упомянутые выше индексы NFS и Fib-4. Несмотря на то что они были разработаны для оценки фиброза у пациентов с заболеваниями печени (в том числе с НАЖБП), они используются и в клинических исследованиях пациентов с СНсФВ. Так, значение индекса Fib-4, соответствующее высокой вероятности выраженного фиброза печени у пациентов исходно без сердечной недостаточности, было ассоциировано с повышенным риском развития СНсФВ, но не сердечной недостаточности со сниженной фракцией выброса [42]. В исследовании 492 пациентов с СНсФВ без первичного поражения печени применялся индекс NFS для оценки взаимосвязи с лабораторными показателями фиброза и прогноза у данной категории пациентов. Интересно, что в этом исследовании лишь у четверти пациентов с СНсФВ значение индекса NFS соответствовало низкому риску развития фиброза печени. Было показано, что увеличение значения индекса NFS ассоциируется с повышением уровня маркеров фиброза и выступает независимым предиктором смерти от всех причин у больных СНсФВ [43].
Следует отметить, что НАЖБП может быть связана с развитием СНсФВ. Так, по данным клинических исследований, наличие у пациента НАЖБП ассоциировано с диастолической дисфункцией левого желудочка [44–46]. Кроме того, поскольку при НАЖБП наблюдается повышение маркеров системного воспаления (СРБ, ФНО-α, ИЛ-6), в том числе в отсутствии СД 2 типа или ожирения, это заболевание само по себе может поддерживать хронический системный воспалительный процесс и быть фактором риска развития СНсФВ [47]. Также одним из возможных механизмов развития СНсФВ у пациентов с НАЖБП при наличии ожирения является появление эктопической жировой ткани в эпикарде сердца. Эпикардиальный жир может выступать источником провоспалительных цитокинов и приводить к нарушению коронарной микроциркуляции, повреждению прилегающего миокарда и его фиброзу [47, 48].
Таким образом, общие патофизиологические процессы (эндотелиальная дисфункция, воспаление, фиброз) указывают на схожесть патогенеза НАЖБП и СНсФВ. Кроме того, развитие СНсФВ может потенциально усугублять течение НАЖБП путем описанных ранее гемодинамических изменений, а хроническое системное воспаление, характерное для НАЖБП, может явиться непосредственной причиной развития диастолической дисфункции левого желудочка и СНсФВ.
МЕХАНИЗМЫ СТРУКТУРНЫХ И ФУНКЦИОНАЛЬНЫХ НАРУШЕНИЙ СКЕЛЕТНОЙ МУСКУЛАТУРЫ ПРИ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ С СОХРАНЕННОЙ ФРАКЦИЕЙ ВЫБРОСА
Один из основных симптомов СНсФВ – плохая переносимость физической нагрузки, определяемая в том числе как снижение пикового потребления кислорода по данным кардиореспираторного тестирования [49, 50]. Помимо изменений сердечной гемодинамики при СНсФВ (фиксированный ударный объем, хронотропная недостаточность), которые могут объяснять снижение физической выносливости у этих пациентов, структурные и функциональные нарушения скелетных мышц могут служить дополнительным фактором плохой переносимости физической нагрузки [51, 52].
Отмечено, что у пациентов с СНсФВ уменьшена мышечная масса, изменен состав мышечных волокон, снижена плотность капилляров, а также нарушен метаболизм скелетных мышц. Так, по данным клинических исследований, при СНсФВ наблюдается снижение общей мышечной массы и мышечной массы ног, измеренных с использованием двухэнергетической рентгеновской абсорбциометрии, и увеличение межмышечной жировой ткани бедра, а также соотношения межмышечного жира к мышечной массе бедра по результатам магнитно-резонансной томографии в сравнении с контрольной группой. При этом изменение данных параметров коррелирует со снижением пикового потребления кислорода при проведении кардиореспираторного тестирования [51, 53].
Гистологические изменения скелетной мышцы включают увеличение числа мышечных волокон типа 2 (гликолитические) с снижением количества мышечных волокон типа 1 (оксидативные), а также уменьшение отношения количества капилляров к мышечным волокнам [54]. Также у больных СНсФВ наблюдается снижение количества митохондрий и активности митохондриальных ферментов, участвующих в окислительном метаболизме [55]. Применение магнитно-резонансной спектроскопии позволило выявить у пациентов с СНсФВ уменьшение времени восстановления энергетических фосфатов (креатинкиназы) после физической нагрузки, более выраженное, нежели у больных СН со сниженной ФВ; это является признаком нарушения метаболических процессов в скелетной мышце у таких пациентов [56]. Данные изменения мышц при СНсФВ можно объяснить снижением градиента перфузии в капиллярах мышечных волокон на фоне фиксированного ударного объема и повышения ЦВД, что может повлечь за собой ишемию, накопление лактата и переключение на гликолитический путь метаболизма глюкозы [57].
Уменьшение мышечной массы в сочетании со снижением силы мышц описывается термином «саркопения», которая встречается у каждого пятого пациента с СНсФВ [7, 58]. Она ассоциирована с повышенным риском падений, ограничением повседневной активности, неспособностью к самообслуживанию [59]. При СНсФВ саркопения играет роль фактора неблагоприятного прогноза, так как увеличивает риск смерти от всех причин в 2,5 раза в течение года после госпитализации по причине декомпенсации сердечной недостаточности [7].
Возрастная дегенерация и связанные с ней гиподинамия, пониженное питание, уменьшение числа мотонейронов мышечных волокон, гормональные изменения (снижение выработки гормона роста, эстрогенов и др.) относятся к основным причинам саркопении [60]. В рамках СНсФВ особый интерес представляет участие в развитии саркопении хронического системного воспаления и эндотелиальной дисфункции.
Результаты клинических исследований свидетельствуют о том, что повышение концентрации провоспалительных цитокинов (ИЛ-6, ФНО-α) и СРБ, характерное для пациентов с саркопенией, связано со снижением мышечной массы и силы мышц – основными проявлениями данного заболевания [61, 62]. Большое значение в запуске и поддержании хронического системного воспалительного процесса в патогенезе саркопении отводится ожирению, что нашло отражение в термине «саркопеническое ожирение» [59, 63]. Несмотря на то что саркопения может вызывать снижение двигательной активности и быть причиной ожирения, существует также мнение, что первоначально при ожирении запускаются патологические процессы, приводящие к потери мышечной массы и снижению функциональной способности скелетной мускулатуры, которые усугубляют метаболические нарушения, замыкая патологический круг [63]. При ожирении возрастание концентрации свободных жирных кислот в крови может приводить к их накоплению в межмышечном пространстве, а также внутри мышечных волокон. Избыточное содержание жирных кислот стимулирует выработку провоспалительных цитокинов (ФНО-α, ИЛ-6 и др.), что запускает патологические процессы, ведущие к дисфункции митохондрий, накоплению активных форм кислорода (оксидативному стрессу) и нарушению противовоспалительных сигнальных путей. Наряду с этим происходит нарушение сигнальный путей рецептора инсулина, в результате чего развивается инсулинорезистентность скелетной мышцы и активизация катаболических процессов в мышечном волокне. В итоге данные процессы ведут к снижению мышечной массы и нарушению функции мышцы, т.е. к саркопении [60, 63].
Эндотелиальная дисфункция и нарушение эндотелийзависимой вазодилатации мышечных сосудов, тесно связанные с хроническим системным воспалением и инсулинорезистентностью мышечных волокон, также относятся к факторам, ассоциированным с развитием саркопении [64]. По данным клинических исследований, дисфункция эндотелия связана со снижением мышечной силы и характерна для пациентов с саркопенией, в том числе страдающих сердечной недостаточностью [65].
В результате структурные и функциональные изменения скелетной мускулатуры и развитие саркопении при СНсФВ могут быть обусловлены не только старением, но и хроническим системным воспалением и эндотелиальной дисфункцией – механизмами, общими для данных заболеваний.
ЗАКЛЮЧЕНИЕ
Пациенты с СНсФВ представляют фенотипически разнородную группу: помимо сердечно-сосудистых заболеваний (АГ, фибрилляция предсердий, легочная гипертензия) при СНсФВ достаточно часто встречаются заболевания почек (ХБП), скелетной мускулатуры (саркопения) и печени (НАЖБП). Представленные механизмы развития этих заболеваний свидетельствуют об общности предрасполагающих факторов и схожести основных звеньев патогенеза с СНсФВ (рис.). При этом взаимосвязь носит двусторонний характер: с одной стороны, СНсФВ путем гемодинамических изменений (повышение ЦВД) может определять развитие внесердечных проявлений, с другой стороны, внесердечные заболевания могут вносить вклад в развитие СНсФВ и усугублять ее симптомы и течение.
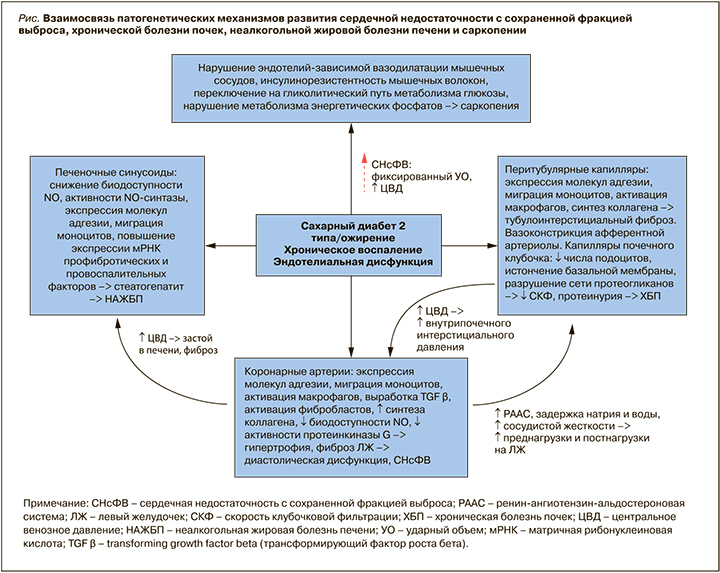
Учитывая участие в патогенезе СНсФВ, ХБП, НАЖБП и саркопении хронического системного воспаления и эндотелиальной дисфункции, а также нарушений углеводного и липидного обмена, можно предположить возможное одновременное поражение соответствующих органов, но с различной по времени манифестацией или с преобладанием патологического процесса в одном органе над другими в силу генетической предрасположенности. Можно ли назвать это звеньями одной цепи? В настоящее время нельзя дать однозначного ответа на этот вопрос, что диктует необходимость дальнейшего изучения данной проблемы. Особенно важно то, что решение этого вопроса может помочь в поиске эффективного метода лечения СНсФВ.


